NF-κB
NF-κB (Nuclear factor kappa-light-chain-enhancer of activated B-cells) zijn menselijke osteoclasten. Het is een familie van transcriptiefactor-eiwitcomplexen die de transcriptie van DNA, cytokineproductie en celoverleving regelen. NF-κB wordt aangetroffen in bijna alle dierlijke celtypen en is betrokken bij cellulaire reacties op stimuli zoals stress, cytokines, vrije radicalen, zware metalen, ultraviolette straling, geoxideerd LDL en bacteriële of virale antigenen.[1][2][3][4][5] NF-κB speelt een sleutelrol bij het reguleren van de immuunrespons op infecties. Onjuiste regulatie van NF-κB is in verband gebracht met kanker, ontstekings- en auto-immuunziekten, septische shock, virusinfectie en onjuiste immuunontwikkeling. NF-κB is ook betrokken bij processen van synaptische plasticiteit en geheugen.[6][7][8][9][10][11]

Ontdekking
NF-κB werd ontdekt door Ranjan Sen in het laboratorium van Nobelprijswinnaar David Baltimore via de interactie ervan met een sequentie van 11 basenparen in de enhancer van de Immunoglobuline lichte keten in B-cellen. Later onderzoek van Alexander Poltorak en Bruno Lemaitre bij muizen en bananenvliegjes stelde Toll-achtige receptoren vast als universeel geconserveerde activatoren van NF-κB-signalering. Deze onderzoeken hebben uiteindelijk bijgedragen aan de toekenning van de Nobelprijs aan Bruce Beutler en Jules A. Hoffmann, die de belangrijkste onderzoekers op dit gebied waren.
Structuur
Alle eiwitten van de NF-κB-familie delen een Rel-homoloogdomein in hun N-terminus. Een subfamilie van NF-κB-eiwitten, waaronder RelA, RelB en c-Rel, hebben een transactivatiedomein in hun C-terminus. Daarentegen worden de NF-κB1- en NF-κB2-eiwitten gesynthetiseerd als grote voorlopers, p105 en p100, die verwerking ondergaan om respectievelijk de p50 (NF-κB1)- en p52 (NF-κB2)-subeenheden te genereren. De werking van p105 en p100 wordt gemedieerd door de ubiquitine/proteasoomroute en omvat selectieve afbraak van hun C-terminusgebied dat ankyrinerepeats bevat. Terwijl het genereren van p52 uit p100 een strak gereguleerd proces is, wordt p50 geproduceerd door constitutieve verwerking van p105. (Van een receptor die in staat is een biologische reactie te veroorzaken in de afwezigheid van een gebonden ligand, wordt gezegd dat deze "constitutieve activiteit" vertoont.) De p50- en p52-eiwitten hebben geen intrinsiek vermogen om transcriptie te activeren en er is daarom voorgesteld dat ze werken als transcriptionele repressoren bij het binden van κB-elementen in homodimeren. Dit verwart inderdaad de interpretatie van p105-knock-outstudies, waarbij door genetische manipulatie een IκB (de volledige lengte van p105) en een waarschijnlijke repressor (p50-homodimeren) en bovendien een transcriptie-activator (de RelA-p50-heterodimeer) is verwijderd.
Familieleden
Leden van de NF-κB-familie delen structureel homologen met het retrovirale oncoproteïne v-Rel, wat resulteert in hun classificatie als NF-κB/Rel-eiwitten.[1].
Er zijn vijf eiwitten in de NF-κB-familie van zoogdieren:[14]
| Klasse | Eiwit | Aliasen | Gen |
|---|---|---|---|
| I | NF-κB1 | p105 → p50 | NFKB1 |
| NF-κB2 | p100 → p52 | NFKB2 | |
| II | RelA | p65 | RELA |
| RelB | RELB | ||
| c-Rel | REL |
De NF-κB/Rel-eiwitten kunnen worden onderverdeeld in twee klassen, die algemene structurele kenmerken delen:[12]

Hieronder staan de lintdiagrammen van vier van de vijf menselijke NF-kB-familieleden:
-
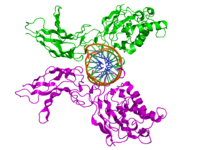 NF-kB1 (p50)
NF-kB1 (p50) -
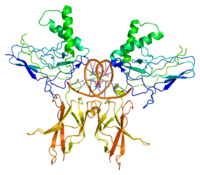 NF-kB2 (p52)
NF-kB2 (p52) -
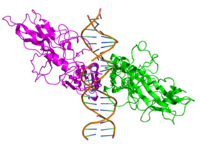 ReLA
ReLA -
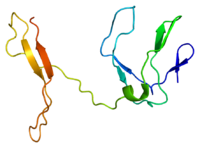 RelB
RelB




Soortendistributie en evolutie
Naast zoogdieren wordt NF-κB ook aangetroffen in een aantal eenvoudige dieren.[15] Deze omvatten neteldieren (zoals zeeanemonen, koraal en hydra), porifera (sponzen), eencellige eukaryoten waaronder Capsaspora owczarzaki en Choanoflagellata, en insecten (zoals motten, muggen en bananenvlieg). De sequentiebepaling van de genomen van de gelekoortsmug, Anopheles gambiae en de bananenvlieg heeft vergelijkende genetische en evolutionaire studies naar NF-κB mogelijk gemaakt. Bij deze insectensoorten wordt NF-κB geactiveerd door de Toll-route (die zich onafhankelijk ontwikkelde bij insecten en zoogdieren) en door de Imd-route (Imd=Immune deficiency).[16]
Signalering
Effect van activering

NF-κB is cruciaal bij het reguleren van cellulaire reacties, omdat het behoort tot de categorie van "snelwerkende" primaire transcriptiefactoren, dat wil zeggen transcriptiefactoren die aanwezig zijn in cellen in een inactieve toestand en geen nieuwe eiwitsynthese vereisen om geactiveerd te worden. (andere leden van deze familie omvatten transcriptiefactoren zoals c-Jun, STATs en kernreceptoren). Hierdoor kan NF-κB een first responder zijn op schadelijke cellulaire stimuli. Bekende inductoren van NF-κB-activiteit zijn zeer variabel en omvatten reactieve zuurstofsoorten (ROS), tumornecrosefactor-alfa (TNFα), interleukine 1-bèta, bacteriële lipopolysachariden (LPS), isoprenaline, cocaïne, endotheline-1 en ioniserende straling.[18]
NF-κB-onderdrukking van cytotoxiciteit van de tumornecrosefactor α (apoptose) is te wijten aan de inductie van antioxiderende enzymen en aanhoudende onderdrukking van c-Jun N-terminus kinasen (JNK's).[19]
Receptoractivator van NF-κB (RANK), een type TNFR, is een centrale activator van NF-κB. Osteoprotegerine (OPG), een lokreceptor-homoloog voor de RANK-ligand (RANKL), remt RANK door te binden aan RANKL, en dus is osteoprotegerine nauw betrokken bij het reguleren van de NF-κB-activering.[20]
Veel bacteriële producten en stimulatie van een grote verscheidenheid aan celoppervlakreceptoren leiden tot NF-κB-activering en vrij snelle veranderingen in genexpressie.[1] De identificatie van Toll-like receptoren (TLR's) als specifieke patroonherkenningsmoleculen en de bevinding dat stimulatie van TLR's leidt tot activering van NF-κB, verbeterde het begrip van hoe verschillende pathogenen NF-κB activeren. Studies hebben bijvoorbeeld TLR4 geïdentificeerd als de receptor voor de lipopolysaccharide-component van gramnegatieve bacteriën.[21] TLR's zijn belangrijke regulatoren van zowel aangeboren als adaptieve immuunreacties.[22]
In tegenstelling tot RelA, RelB en c-Rel bevatten de p50- en p52-NF-κB-subeenheden geen transactivatiedomeinen in hun C-terminushelften.[23] Niettemin spelen de p50- en p52-NF-κB-leden een cruciale rol bij het moduleren van de specificiteit van de NF-κB-functie. Hoewel homodimeren van p50 en p52 in het algemeen repressoren zijn van de transcriptie van de κB-plaats, nemen zowel p50 als p52 deel aan de transactivatie van het doelgen door heterodimeren te vormen met RelA, RelB of c-Rel. Bovendien binden p50- en p52-homodimeren ook aan het kerneiwit BCL3 en dergelijke complexen kunnen functioneren als transcriptionele activatoren.[24][25][26]
Remming
In niet-gestimuleerde cellen worden de NF-κB-dimeren in het cytoplasma afgezonderd door een familie van remmers, genaamd IκB's (remmer van κB ), dit zijn eiwitten die meerdere repeats bevatten van een sequentie die ankyrinerepetas wordt genoemd. Dankzij hun ankyrine-repeatdomeinen maskeren de IκB-eiwitten de kernlokaliseringssignalen (NLS) van NF-κB-eiwitten en houden ze ze in een inactieve toestand in het cytoplasma opgesloten.[27]
IκB's zijn een familie van verwante eiwitten die een N-terminusregulerend domein hebben, gevolgd door zes of meer ankyrinerepeats en een PEST-domein nabij hun C-terminus. Hoewel de IκB-familie bestaat uit IκBα, IκBβ, IκBε en BCL3, is IκBα het best bestudeerde en belangrijkste IκB-eiwit. Vanwege de aanwezigheid van ankyrinerepeats in hun C-terminus helften functioneren p105 en p100 ook als IκB-eiwitten. De C-terminus helft van p100, die vaak IκBδ wordt genoemd, functioneert ook als remmer.[28][29] De afbraak van IκBδ als reactie op ontwikkelingsstimuli, zoals die getransduceerd via LTβR, versterkt de activering van NF-κB-dimeer in een NIK (NF-kappa-B-inducing kinase)-afhankelijke niet-canonieke (niet=gebruikelijke) route.[28][30]
Activeringsproces (canoniek/klassiek)
Activering van NF-κB wordt geïnitieerd door de signaalgeïnduceerde afbraak van IκB-eiwitten. Dit gebeurt voornamelijk via activering van het IκB-kinase (IKK). IKK is samengesteld uit een heterodimeer van de katalytische IKKα- en IKKβ-subeenheden en een regulerend eiwit genaamd NEMO (NF-κB essentiële modulator) of IKKγ. Wanneer geactiveerd door signalen, meestal afkomstig van buiten de cel, fosforyleert het IκB-kinase twee serineresiduen die zich in een IκB-regulerend domein bevinden. Wanneer ze op deze serinen worden gefosforyleerd (bijvoorbeeld serinen 32 en 36 in menselijke IκBα), worden de IκB-eiwitten gemodificeerd door een proces dat ubiquitinatie wordt genoemd, wat er vervolgens toe leidt dat ze worden afgebroken door het proteasoom.
Met de afbraak van IκB komt het NF-κB-complex vrij en kan het de celkern binnen dringen, waar het de expressie kan 'aanzetten' van specifieke genen die DNA-bindingsplaatsen voor NF-κB in de buurt hebben. De activering van deze genen door NF-κB leidt vervolgens tot de gegeven fysiologische respons, bijvoorbeeld een ontstekings- of immuunrespons, een celoverlevingsrespons of cellulaire proliferatie. Translocatie van NF-κB naar de celkern kan immunocytochemisch worden gedetecteerd en gemeten met laserscanningcytometrie.[31] NF-κB schakelt de expressie van zijn eigen repressor, IκBα, in. Het nieuw gesynthetiseerde IκBα remt vervolgens NF-κB opnieuw en vormt zo een automatische feedbacklus, wat resulteert in oscillerende niveaus van NF-κB-activiteit.[32] Bovendien hebben verschillende virussen, waaronder het AIDS-virus Hiv, bindingsplaatsen voor NF-κB die de expressie van virale genen controleren, die op hun beurt bijdragen aan virale replicatie of virale pathogeniteit. In het geval van Hiv-1 kan de activering van NF-κB, tenminste gedeeltelijk, betrokken zijn bij de activering van het virus vanuit een latente, inactieve toestand.[33] YopP is een factor die wordt uitgescheiden door Yersinia pestis, de veroorzaker van de pest, en die de alomtegenwoordigheid van IκB voorkomt. Dit zorgt ervoor dat deze ziekteverwekker de NF-κB-route effectief remt en zo de immuunrespons bij een door Yersinia pestisgeïnfecteerde mens blokkeert.[34]
Remmers van NF-KB-activiteit
IFRD1 (Interferon-related developmental regulator 1) is een van de bekende eiwitremmers van de NF-κB-activiteit en onderdrukt de activiteit van NF-κB p65 door de HDAC (histone deacetylase)-gemedieerde deacetylering van de p65-subeenheid op lysine 310 te versterken, door de rekrutering van HDAC3 naar p65 te bevorderen. In feite vormt IFRD1 trimoleculaire complexen met p65 en HDAC3.[35][36]
De NAD+-afhankelijke proteïnedeacetylase en levensduurfactor SIRT1 (silent mating type information regulation 2 homolog) 1) remt de genexpressie van NF-κB door de RelA/p65-subeenheid van NF-κB op lysine 310 te deacetyleren.[37]
Niet-canonieke/alternatieve route
Een selecte reeks celdifferentiërende of ontwikkelingsstimuli, zoals lymfotoxine β-receptor (LTβR), BAFF (B-cell activating factor) of RANKL (receptor activator of nuclear factor kappa-Β ligand), activeren de niet-canonieke NF-κB-route om NF-κB/RelB:p52-dimeer in de celkern te induceren. In deze route leidde activering van het NF-κB-inducerende kinase (NIK) na receptorligatie tot de fosforylatie en daaropvolgende proteasomale verwerking van het NF-κB2-precursoreiwit p100 tot volwassen p52-subeenheid op een IKK1/IKKa-afhankelijke manier. Vervolgens dimeriseert p52 met RelB voor een nucleaire RelB:p52 DNA-bindende activiteit. RelB:p52 reguleert de expressie van homeostatische lymfokinen, die de lymfoïde organogenese en lymfocytenhandel in de secundaire lymfoïde organen instrueren.[38] In tegenstelling tot de canonieke signalering die afhankelijk is van door NEMO-IKK2 gemedieerde afbraak van IκBα, -β, -ε, hangt niet-canonieke signalering af van door NIK gemedieerde omzetting van p100 naar p52. Gezien hun verschillende reguleringen werd aangenomen dat deze twee routes onafhankelijk van elkaar waren. Er werd echter gevonden dat de synthesen van de bestanddelen van de niet-canonieke route, namelijk RelB en p52, worden gecontroleerd door canonieke IKK2-IκB-RelA:p50-signalering.[39] Bovendien is de generatie van de canonieke en niet-canonieke dimeren, namelijk RelA:p50 en RelB:p52, binnen het cellulaire milieu mechanisch met elkaar verbonden.[39] Deze analyses suggereren dat een geïntegreerd NF-κB-systeemnetwerk ten grondslag ligt aan de activering van zowel RelA als RelB-bevattend dimeer en dat een slecht functionerende canonieke route ook via de niet-canonieke route tot een afwijkende cellulaire respons zal leiden. Het meest intrigerend is dat een recente studie heeft aangetoond dat door TNF geïnduceerde canonieke signalering de niet-canonieke RelB:p52-activiteit in de ontstoken lymfoïde weefsels ondermijnt, waardoor het binnendringen van lymfocyten wordt beperkt.[40] Mechanisch gezien inactiveerde TNF NIK in door LTβR gestimuleerde cellen en induceerde de synthese van Nfkb2 (Nuclear factor NF-kappa-B p100 subunit )-mRNA dat codeert voor p100; deze samen accumuleerden onverwerkt p100, wat de RelB-activiteit verzwakte. Een rol van p100/Nfkb2 bij het dicteren van het binnendringen van lymfocyten in het ontstoken lymfoïde weefsel kan brede fysiologische implicaties hebben.
Naast zijn traditionele rol in de lymfoïde organogenese, versterkt de niet-canonieke NF-κB-route ook direct de inflammatoire immuunreacties op microbiële pathogenen door de canonieke NF-κB-signalering te moduleren. Er werd aangetoond dat p100/Nfkb2 stimulusselectieve en celtypespecifieke overspraak tussen de twee NF-κB-routes medieert en dat door Nfkb2 gemedieerde overspraak muizen beschermt tegen darmpathogenen. Aan de andere kant herpositioneert een gebrek aan door p100 gemedieerde regelingen RelB onder de controle van door TNF geïnduceerde canonieke signalering. In feite stelde mutatie-inactivatie van p100/Nfkb2 bij een myeloom TNF in staat een langdurige RelB-activiteit te induceren, die zorgde voor resistentie in plasmacellen tegen chemotherapeutische geneesmiddelen.[41]
Bij immuniteit
NF-κB is een belangrijke transcriptiefactor die genen reguleert die verantwoordelijk zijn voor zowel de aangeboren als de adaptieve immuunrespons.[42] Na activering van de T- of B-celreceptor wordt NF-κB geactiveerd via verschillende signaalcomponenten. Na ligatie van de T-celreceptor wordt proteïnekinase Lck (lymphocyte-specific protein tyrosine kinase) gerekruteerd en fosforyleert de ITAM's (Immunoreceptor tyrosine-based activation motif) van de CD3-cytoplasmatische staart. ZAP70 (Zeta-chain-associated protein kinase 70) wordt vervolgens gerekruteerd in de gefosforyleerde ITAM's en helpt bij het rekruteren van LAT (Linker for activation of T cells) en PLC-γ (1-Phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2), wat activering van PKC (protein kinase C) veroorzaakt. Door een cascade van fosforyleringsgebeurtenissen wordt het kinasecomplex geactiveerd en kan NF-κB de celkern binnendringen om genen die betrokken zijn bij de ontwikkeling, rijping en proliferatie van T-cellen up te reguleren.[43]
In het zenuwstelsel
Naast een rol bij het bemiddelen in de overleving van cellen, hebben onderzoeken van Mark Mattson en anderen aangetoond dat NF-κB diverse functies in het zenuwstelsel vervult, waaronder rollen in synaptische plasticiteit, leren en geheugen.[44] Naast stimuli die NF-κB in andere weefsels activeren, kan NF-κB in het zenuwstelsel worden geactiveerd door groeifactoren (BDNF, zenuwgroeifactor (NGF)) en synaptische transmissie zoals glutamaat.[7] Deze activatoren van NF-κB in het zenuwstelsel komen allemaal samen in het IKK-complex en de canonieke route.
Er is de laatste tijd veel belangstelling voor de rol van NF-κB in het zenuwstelsel. Huidige studies suggereren dat NF-κB belangrijk is voor het leren en het geheugen van meerdere organismen, waaronder krabben,[9][10] fruitvliegjes[45] en muizen.[7][8] NF-κB kan het leren en het geheugen gedeeltelijk reguleren door de synaptische plasticiteit[6][46] en de synapsfunctie[45][47][48] te moduleren en door de groei van dendrieten[49] en dendritische spines te reguleren.[48]
Van genen met NF-κB-bindingsplaatsen is aangetoond dat ze na het leren een verhoogde expressie hebben,[8] wat suggereert dat de transcriptiedoelen van NF-κB in het zenuwstelsel belangrijk zijn voor plasticiteit. Veel NF-κB-doelgenen die belangrijk kunnen zijn voor plasticiteit en leren omvatten groeifactoren (BDNF, NGF),[50] cytokinen (tumornecrosefactor α (TNF-α), TNFR (tumor necrosis factor receptor))[51] en kinasen (PKAc) (protein kinase A).[46]
Ondanks het functionele bewijs voor een rol voor transcriptiefactoren uit de Rel-familie in het zenuwstelsel, is het nog steeds niet duidelijk dat de neurologische effecten van NF-κB de transcriptionele activering in zenuwcellen weerspiegelen. De meeste manipulaties en testen worden uitgevoerd in de gemengde celomgevingen die in vivo worden aangetroffen, in "neuronale" celculturen die aanzienlijke aantallen gliacellen bevatten, of in van tumoren afgeleide "neuronale" cellijnen. Wanneer transfecties of andere manipulaties specifiek op zenuwcellen zijn gericht, zijn de gemeten eindpunten doorgaans elektrofysiologie of andere parameters die helemaal niet lijken op transcriptie. Zorgvuldige tests van NF-κB-afhankelijke transcriptie in sterk gezuiverde zenuwcelculturen vertonen over het algemeen weinig tot geen NF-κB-activiteit.[52][53]
Sommige meldingen van NF-κB in zenuwcellen lijken een artefact te zijn geweest van niet-specificiteit van antilichamen..[54] Natuurlijk kunnen artefacten van een celcultuur – bijvoorbeeld door zenuwcellen te onttrekken aan de invloed van gliacellen – ook valse resultaten opleveren. Maar dit is aangepakt in ten minste twee co-cultuurbenaderingen. Moerman et al.[55] gebruikten een co-cultuurformat waarbij zenuwcellen en gliacellen na behandeling konden worden gescheiden voor EMSA (electrophoretic mobility shift assay)-analyse, en ze ontdekten dat de NF-κB geïnduceerd door glutamaatstimuli beperkt was tot gliacellen (en, intrigerend genoeg, alleen gliacellen die 48 uur in de aanwezigheid van zenuwcellen waren geweest). Dezelfde onderzoekers onderzochten het probleem met een andere benadering, waarbij gebruik werd gemaakt van zenuwcellen van een transgene NF-κB-reportermuis gekweekt met wildtype gliacellen; glutamaatstimuli konden opnieuw niet worden geactiveerd in zenuwcellen.[56] Een deel van de DNA-bindende activiteit die onder bepaalde omstandigheden wordt opgemerkt (met name die gerapporteerd als constitutief) lijkt het gevolg te zijn van Sp3 (Sp3-transcriptiefactor)- en Sp4-binding aan een subset van KB-enhancersequenties in zenuwcellen.[57] Deze activiteit wordt feitelijk geremd door glutamaat en andere aandoeningen die het calcium in de zenuwcel verhogen. Uiteindelijk blijft de rol van NF-κB in zenuwcellen ondoorzichtig vanwege de moeilijkheid om de transcriptie te meten in cellen die tegelijkertijd op type worden geïdentificeerd. Het leren en het geheugen kunnen zeker worden beïnvloed door transcriptionele veranderingen in astrocyten en andere gliacel-elementen. En er moet rekening mee worden gehouden dat er naast de directe transactivatie (verhoogde snelheid van genexpressie) van genen ook mechanische effecten van NF-κB kunnen zijn.
Klinische betekenis
NF-κB wordt veel gebruikt door eukaryote cellen als regulator van genen die celproliferatie en celoverleving controleren. Als zodanig hebben veel verschillende soorten menselijke tumoren een verkeerd gereguleerd NF-κB: dat wil zeggen dat NF-κB constitutief actief is. Actieve van NF-κB zet de expressie aan van genen die ervoor zorgen dat de cel blijft prolifereren en de cel beschermt tegen omstandigheden die er anders voor zouden zorgen dat de cel zou afsterven via apoptose. Bij kanker zijn eiwitten die de NF-κB-signalering controleren gemuteerd of op een afwijkende manier tot expressie gebracht, wat leidt tot een gebrekkige coördinatie tussen de kwaadaardige cel en de rest van het organisme. Dit blijkt zowel uit metastase als uit de inefficiënte uitroeiing van de tumor door het immuunsysteem.[58]
Normale cellen kunnen dood gaan wanneer ze worden verwijderd uit het weefsel waartoe ze behoren, of wanneer hun genoom niet in harmonie met de weefselfunctie kan functioneren: deze gebeurtenissen zijn afhankelijk van feedbackregulatie van NF-κB en falen bij kanker.[59]
Defecten in NF-κB resulteren in een verhoogde gevoeligheid voor apoptose, wat leidt tot verhoogde celdood. Dit komt omdat NF-κB anti-apoptotische genen reguleert, vooral de TRAF1 (TNF receptor-associated factor 1) en TRAF2, en daarom de activiteiten van de caspase-familie van enzymen opheft, die centraal staan in de meeste apoptotische processen.[60]

In tumorcellen is de NF-κB-activiteit verhoogd, zoals bijvoorbeeld bij 41% van de gevallen van nasofarynxcarcinoom,[61] darmkanker, prostaatkanker en alvleeskliertumoren. Dit is ofwel het gevolg van mutaties in genen die coderen voor de NF-κB-transcriptiefactoren zelf of in genen die de NF-κB-activiteit controleren (zoals IκB (IκB kinase)-genen); Bovendien scheiden sommige tumorcellen stoffen uit die ervoor zorgen dat NF-κB actief wordt.[62][63] Het blokkeren van NF-κB kan ervoor zorgen dat tumorcellen stoppen met prolifereren, afsterven of gevoeliger worden voor de werking van chemotherapeutische middelen.[64][65] Zo is NF-κB het onderwerp van veel actief onderzoek onder farmaceutische bedrijven voor chemotherapie[66] en kankerimmunotherapie.
Hoewel overtuigende experimentele gegevens NF-κB hebben geïdentificeerd als een kritische promotor van kankervorming, wat een goede reden geeft voor de ontwikkeling van een antitumortherapie die is gebaseerd op onderdrukking van NF-κB-activiteit, moet echter voorzichtigheid worden betracht bij het overwegen van een anti-NF-κB-therapie als een brede therapeutische strategie bij de behandeling van kanker, aangezien gegevens ook hebben aangetoond dat NF-κB -activiteit de gevoeligheid van tumorcellen voor apoptose en veroudering verhoogt. Bovendien is aangetoond dat de canonieke NF-κB een Fas-transcriptie-activator is en dat de alternatieve NF-κB een Fas-transcriptierepressor is.[67] Daarom bevordert NF-κB Fas-gemedieerde apoptose in kankercellen en dus kan remming van NF-κB Fas-gemedieerde apoptose onderdrukken en zo de door immuuncellen gemedieerde tumoronderdrukking van de gastheer negatief beïnvloeden.
Ontsteking
Omdat NF-κB veel genen controleert die betrokken zijn bij ontstekingen, is het niet verrassend dat NF-κB chronisch actief blijkt te zijn bij veel ontstekingsziekten, zoals inflammatoire darmziekte, artritis, sepsis, gastritis, astma, atheromatose[68] en andere. Een verhoging van sommige NF-κB-activatoren, zoals osteoprotegerine (OPG), is geassocieerd met een verhoogde mortaliteit, vooral als gevolg van hart- en vaatziekten..[69][70] Verhoogde NF-κB is ook in verband gebracht met schizofrenie.[71] Onlangs is NF-κB-activering voorgesteld als een mogelijk moleculair mechanisme voor de katabole effecten van sigarettenrook in skeletspieren en sarcopenie.[72] Onderzoek heeft aangetoond dat tijdens ontstekingen de functie van een cel afhangt van signalen die deze activeert als reactie op contact met aangrenzende cellen en op combinaties van hormonen, vooral cytokines die erop inwerken via specifieke receptoren.[73] Het fenotype van een cel in een weefsel ontwikkelt zich door wederzijdse stimulatie van terugkoppelingsignalen die de functie ervan met andere cellen coördineren. Dit is vooral duidelijk tijdens het herprogrammeren van de celfunctie wanneer een weefsel wordt blootgesteld aan ontstekingen, omdat cellen hun fenotype veranderen en geleidelijk combinaties van genen tot expressie brengen die het weefsel voorbereiden op regeneratie nadat de oorzaak van de ontsteking is weggenomen.[73][74] Bijzonder belangrijk zijn de terugkoppelingsreacties die zich ontwikkelen tussen in het weefsel blijven zittende cellen en circulerende cellen van het immuunsysteem.[74]
De betrouwbaarheid van terugkoppelingsreacties tussen diverse celtypen en het immuunsysteem hangt af van de integriteit van mechanismen die het bereik van door NF-κB geactiveerde genen beperken, waardoor alleen de expressie mogelijk is van genen die bijdragen aan een effectieve immuunrespons en vervolgens aan een volledig herstel van het functioneren van het weefsel na het verdwijnen van de ontsteking.[74] Bij kanker zijn de mechanismen die de genexpressie reguleren als reactie op ontstekingsstimuli zo veranderd dat een cel zijn overleving niet langer koppelt aan de mechanismen die zijn fenotype en zijn functie coördineren met de rest van het weefsel.[59] Dit komt vaak tot uiting in een ernstig gecompromitteerde regulatie van NF-κB-activiteit, waardoor kankercellen abnormale groepen van NF-κB-doelgenen tot expressie kunnen brengen.[75] Dit heeft tot gevolg dat niet alleen de kankercellen abnormaal gaan functioneren: de cellen van het omringende weefsel veranderen hun functie en ondersteunen niet langer uitsluitend het organisme. Bovendien kunnen verschillende soorten cellen in de micro-omgeving van kanker hun fenotypes veranderen om de groei van kanker te ondersteunen.[76][77][78] Ontsteking is daarom een proces dat de betrouwbaarheid van weefselcomponenten test, omdat het proces dat tot weefselregeneratie leidt coördinatie van genexpressie tussen verschillende celtypen vereist.[73][79]
NEMO
Het NEMO-deficiëntiesyndroom is een zeldzame genetische aandoening die verband houdt met een fout in de IKBKG (inhibitor of nuclear factor kappa-B kinase subunit gamma) die op zijn beurt NF-κB activeert. Het treft vooral mannen en heeft een zeer wisselende reeks symptomen en prognoses.[80]
Veroudering en obesitas
NF-κB komt steeds meer tot expressie bij obesitas en veroudering, [91] resulterend in verlaagde niveaus van het ontstekingsremmende, pro-autofagie, anti-insulineresistentie-eiwit sirtuin 1. NF-κB verhoogt de niveaus van het microRNA miR-34a (dat de NAD-synthese remt) door te binden aan zijn promotorregio resulterend in lagere niveaus van sirtuin 1.
NF-κB en interleukine 1α stimuleren elkaar wederzijds in verouderende cellen in een positieve terugkoppelingslus, waardoor de verouderingsgeassocieerde secretoire fenotype (SASP) factoren worden aangemaakt. NF-κB en het nicotinamide-adenine-dinucleotide-afbrekende enzym CD38 (cyclisch ADP-ribosehydrolase) stimuleren elkaar ook wederzijds.
NF-κB is een centraal onderdeel van de cellulaire reactie op schade. NF-κB wordt geactiveerd in verschillende celtypen die normale of versnelde veroudering ondergaan. Genetische of farmacologische remming van NF-κB-activering kan het begin van talrijke verouderingsgerelateerde symptomen en pathologieën vertragen. Dit effect kan gedeeltelijk worden verklaard doordat de reductie van NF-κB de productie van van mitochondriën afkomstige reactieve zuurstofcomponenten vermindert die DNA kunnen beschadigen.
Bronnen, noten en/of referenties
|










